- Definition
- Evaluation Numerical gradients Analytic gradient methods
- Crossings and avoided crossings of potential energy surfaces
- Difficulties and alternatives
- Theoretical development
- See also
- References
Vibronic coupling (also called nonadiabatic coupling or derivative coupling) in a molecule involves the interaction between electronic and nuclear vibrational motion.[1][2] The term "vibronic" originates from the combination of the terms "vibrational" and "electronic", denoting the idea that in a molecule, vibrational and electronic interactions are interrelated and influence each other. The magnitude of vibronic coupling reflects the degree of such interrelation.
In theoretical chemistry, the vibronic coupling is neglected within the Born–Oppenheimer approximation. Vibronic couplings are crucial to the understanding of nonadiabatic processes, especially near points of conical intersections.[3][4] The direct calculation of vibronic couplings is not common due to difficulties associated with its evaluation.
Definition
Vibronic coupling describes the mixing of different electronic state as a result of small vibrations.
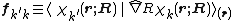
Evaluation
The evaluation of vibronic coupling often involve complex mathematical treatment.
Numerical gradients
The form of vibronic coupling is essentially derivative of the wave function. Each component of the vibronic coupling vector can be calculated with numerical differentiation methods using wave functions at displaced geometries. This is the procedure used in MOLPRO.[5]
First order accuracy can be achieved with forward difference formula:
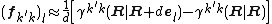
Second order accuracy can be achieved with central difference formula:
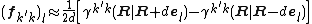
Here, 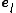 is a unit vector along direction
is a unit vector along direction  .
. 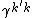 is the transition density between the two electronic states.
is the transition density between the two electronic states.
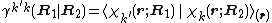
Evaluation of electronic wave functions for both electronic states are required at N displacement geometries for first order accuracy and 2*N displacements to achieve second order accuracy, where N is the number of nuclear degrees of freedom. This can be extremely computationally demanding for large molecules.
As with other numerical differentiation method, the evaluation of nonadiabatic coupling vector with this method is numerically unstable, limiting the accuracy of the result. Moreover, the calculation of the two transition densities in the numerator are not straightforward. The wave functions of both electronic states are expanded with Slater determinants or Configuration state functions (CSF). The contribution from the change of CSF basis is too demanding to evaluate using numerical method, and is usually ignored by employing an approximate diabatic CSF basis. This will also cause further inaccuracy of the calculated coupling vector, although this error is usually tolerable.
Analytic gradient methods
Evaluating derivative couplings with analytic gradient methods has the advantage of high accuracy and very low cost, usually much cheaper than one single point calculation. This means an acceleration factor of 2N. However, the process involves intense mathematical treatment and programming. As a result, few programs have currently implemented analytic evaluation of vibronic couplings. Details about this method can be found in ref.[6] For the implementation for SA-MCSCF and MRCI in COLUMBUS, please see ref.[7]
Crossings and avoided crossings of potential energy surfaces
{{Main|Potential energy surface|Conical intersection}}Vibronic coupling is large in the case of two adiabatic potential energy surfaces coming close to each other (that is, when the energy gap between them is of the order of magnitude of one oscillation quantum). This happens in the neighbourhood of an avoided crossing of potential energy surfaces corresponding to distinct electronic states of the same spin symmetry. At the vicinity of conical intersections, where the potential energy surfaces of the same spin symmetry cross, the magnitude of vibronic coupling approaches infinity. In either case the adiabatic or Born–Oppenheimer approximation fails and vibronic couplings have to be taken into account.
The large magnitude of vibronic coupling near avoided crossings and conical intersections allows wave functions to propagate from one adiabatic potential energy surface to another, giving rise to nonadiabatic phenomena such as radiationless decay. The singularity of vibronic coupling at conical intersections is responsible for the existence of Berry phase, which has been discovered by Longuet-Higgins in this context.
Difficulties and alternatives
Although crucial to the understanding of nonadiabatic processes, direct evaluation of vibronic couplings has been very limited.
Evaluation of vibronic couplings is often associated with severe difficulties in mathematical formulation and program implementations. As a result, the algorithms to evaluate vibronic couplings are not yet implemented in many quantum chemistry program suites.
The evaluation of vibronic couplings also requires correct description of at least two electronic states in regions where they are strongly coupled. This requires the use of multi-reference methods such as MCSCF and MRCI, which are computationally demanding and delicate quantum-chemical methods. This is further complicated by the fact that definition of vibronic couplings requires electronic wave functions. Unfortunately, wave function based methods are usually too expensive for larger systems and popular methods for larger systems such as density functional theory and molecular mechanics cannot generate wave function information.
As a result, direct evaluation of vibronic couplings are mostly limited to very small molecules. The magnitude of vibronic coupling is often introduced as an empirical parameter determined by reproducing experimental data.
Alternatively, one can avoid explicit use of derivative couplings by switch from the adiabatic to the diabatic representation of the potential energy surfaces. Although rigorous validation of a diabatic representation requires knowledge of vibronic coupling, it is often possible to construct such diabatic representations by referencing the continuity of physical quantities such as dipole moment, charge distribution or orbital occupations. However, such construction requires detailed knowledge of a molecular system and introduces significant arbitrariness. Diabatic representations constructed with different method can yield different results and the reliability of the result relies on the discretion of the researcher.
Theoretical development
Perhaps the earliest examples of the importance of vibronic coupling were found during the 1930s. In 1934 Renner wrote about the vibronic coupling in an electronically excited Π-state in CO2. Calculations of the lower excited levels of benzene by Sklar in 1937 (with the valence bond method) and later in 1938 by Goeppert-Mayer and Sklar (with the molecular orbital method) demonstrated a correspondence between the theoretical predictions and experimental results of the benzene spectrum. The benzene spectrum was the first qualitative computation of the efficiencies of various vibrations at inducing intensity absorption.[8]
See also
- Born–Huang approximation
- Born–Oppenheimer approximation
- Conical intersection
References
2. ^{{ cite journal|doi=10.1111/j.1751-1097.1977.tb06918.x |first=T. |last=Azumi|title=WHAT DOES THE TERM "VIBRONIC COUPLING" MEAN?|journal= Photochemistry and Photobiology|volume= 25|issue=3 | pages=315–326|year=1977}}
3. ^{{cite journal|last=Yarkony|first=David R.|title=Nonadiabatic Quantum Chemistry—Past, Present, and Future|journal=Chemical Reviews|date=11 January 2012|volume=112|issue=1|pages=481–498|doi=10.1021/cr2001299|pmid=22050109}}
4. ^{{cite book|last=Baer|first=Michael|title=Beyond Born-Oppenheimer : electronic non-adiabatic coupling terms and conical intersections|year=2006|publisher=Wiley|location=Hoboken, N.J.|isbn=978-0471778912}}
5. ^{{cite web|title=NON ADIABATIC COUPLING MATRIX ELEMENTS|url=http://www.molpro.net/info/2012.1/doc/manual/node509.html|publisher=MOLPRO|accessdate=3 November 2012}}
6. ^{{cite journal|last=Lengsfield|first=Byron H.|author2=Saxe, Paul |author3=Yarkony, David R. |title=On the evaluation of nonadiabatic coupling matrix elements using SA-MCSCF/CI wave functions and analytic gradient methods. I|journal=The Journal of Chemical Physics|date=1 January 1984|volume=81|issue=10|pages=4549|doi=10.1063/1.447428|bibcode = 1984JChPh..81.4549L }}
7. ^{{cite journal|last=Lischka|first=Hans|author2=Dallos, Michal |author3=Szalay, Péter G. |author4=Yarkony, David R. |author5= Shepard, Ron |title=Analytic evaluation of nonadiabatic coupling terms at the MR-CI level. I. Formalism|journal=The Journal of Chemical Physics|date=1 January 2004|volume=120|issue=16|pages=7322|doi=10.1063/1.1668615|pmid=15267642|bibcode = 2004JChPh.120.7322L }}
8. ^{{cite book |last=Fischer |first=Gad |title=Vibronic Coupling: The Interaction between the Electronic and Nuclear Motions |publisher=Academic Press |location=New York |year=1984 |isbn=978-0-12-257240-1 }}
- Cinquefoil tortrix
- Cinquefoil Twist
- Cinquefoil twist
- Cinque Hommes Creek (stream), Perry County, Missouri
- Cinque Port constituency
- Cinque Port Light Dragoons
- Cinque Ports Light Dragoons
- Cinque Ports Light Railway
- Cinquetti
- Cinquil
- Cinquini
- Cinquo de MAyo
- Cin-saan
- Cinsia Savi Scarponi
- C. insignis
- Cinta Ilahi
- Cinta Kita
- Cinta Larga people
- Cinta Laura Kiehl
- Cinta Pertama (film)
- Cintapro
- Cintas Fellowship
- Cinteteo
- Cinthia Knoth
- Cinthia Martinez