- Radiation exposure in everyday life Radiation and medical imaging
- Target theory The basic ballistic models The single-target single-hit model The n-target single-hit model The linear quadratic model The three lambda model The linear-quadratic-cubic model Sublesions hypothesis models The repair-misrepair model The lethal-potentially lethal model The saturable repair model
- Cellular environment and radiation hormesis Radiation hormesis Radiation-Induced Bystander Effect (RIBE)
- Radiation and oxidative stress Formation of ROS Activation of NOS Mechanism of oxidative stress and epigenetic gene regulation ROS-mediated protein adduct formation ROS-mediated DNA methylation changes ROS-mediated post-translation modification ROS-mediated loss of epigenetic imprinting
- Mechanisms of epigenetic modifications Ionizing radiation and DNA methylation Genomic instability via hypomethylation of LINE1 Ionizing radiation and histone modification Loss of methylation via repair mechanisms
- Clinical consequences and applications Epigenetic affects on a developing brain MGMT- and LINE1- specific DNA methylation Treatment by DNMT inhibitors
- References
Ionizing radiation has been known to cause damage to cellular components such as proteins, lipids, and nucleic acids. It has also been known to cause DNA double-strand breaks. Accumulation of DNA double strand breaks can lead to cell cycle arrest in somatic cells and cause cell death. Due to its ability to induce cell cycle arrest, ionizing radiation is used on abnormal growths in the human body such as cancer cells, in radiation therapy. Most cancer cells are fully treated with some type of radiotherapy, however some cells such as stem cell cancer cells show a reoccurrence when treated by this type of therapy.[1]
Radiation exposure in everyday life
Non-ionising radiations, electromagnetic fields (EMF) such as radiofrequency (RF), or power frequency radiation have become very common in everyday life. All of these exist as low frequency radiation which can come from wireless cellular devices or through electrical appliances which induce extremely low frequency radiation (ELF). Exposure to these radioactive frequencies has shown negative affects on the fertility of men by impacting the DNA of the sperm and deteriorating the testes[2] as well as an increased risk of tumor formation in salivary glands.[3][4] The International Agency for Research on Cancer considers RF electromagnetic fields to be possibly carcinogenic to humans, however the evidence is limited.[5]
Radiation and medical imaging
{{see also|Radiation protection}}Advances in medical imaging has resulted in increased exposure of humans to low doses of ionizing radiation. Radiation exposure in pediatrics has been shown to have a greater impact as children's cells are still developing.[2] The radiation obtained from medical imaging techniques is only harmful if consistently targeted multiple times in a short space of time. Safety measures have been introduced in order to limit the exposure of harmful ionizing radiation such as the usage of protective material during the use of these imaging tools. A lower dosage is also used in order to fully rid the possibility of a harmful effect from the medical imaging tools. The National Council on Radiation Protection and Measurements along with many other scientific committees have ruled in favor of continued use of medical imaging as the reward far outweighs the minimal risk obtained from these imaging techniques. If the safety protocols are not followed there is a potential increase in the risk of developing cancer. This is primarily due to the decreased methylation of cell cycle genes, such as those relating to apoptosis and DNA repair. The ionizing radiation from these techniques can cause many other detrimental effects in cells including changes in gene expression and halting the cell cycle. However, these results are extremely unlikely if the proper protocols are followed.[1][4]
Target theory
Target theory concerns the models of how radiation kills biological cells and is based around two main postulates:
- "Radiation is considered to be a sequence of random projectiles;
- the components of the cell are considered as the targets bombarded by these projectiles"&91;6&93;
Several models have been based around the above two points. From the various proposed models three main conclusions were found:
- Physical hits obey a Poisson distribution
- Failure of radioactive particles to attack sensitive areas of cells allow for survival of the cell
- Cell death is an exponential function of the dose of radiation received as the number of hits received is directly proportional to the radiation dose; all hits are considered lethal&91;7&93;
Radiation exposure through ionizing radiation (IR) affects a variety of processes inside of an exposed cell. IR can cause changes in gene expression, disruption of cell cycle arrest, and apoptotic cell death. The extent of how radiation effects cells depends on the type of cell and the dosage of the radiation. Some irradiated cancer cells have been shown to exhibit DNA methylation patterns due to epigenetic mechanisms in the cell. In medicine, medical diagnostic methods such as CT scans and radiation therapy expose the individual to ionizing radiation. Irradiated cells can also induce genomic instability in neighboring un-radiated cells via the bystander effect. Radiation exposure could also occur via many other channels than just ionizing radiation.
The basic ballistic models
The single-target single-hit model
In this model a single hit on a target is sufficient to kill a cell[7] The equation used for this model is as follows:
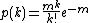
Where k represents a hit on the cell and m represents the mass of the cell.
The n-target single-hit model
In this model the cell has a number of targets n. A single hit on one target is not sufficient to kill the cell but does disable the target. An accumulation of successful hits on various targets leads to cell death.[7] The equation used for this model is as follows:
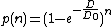
Where n represents number of the targets in the cell.
The linear quadratic model
The equation used for this model is as follows:[7]
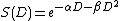
where αD represents a hit made by a one particle track and βD represents a hit made by a two particle track and S(D) represents the probability of survival of the cell.
The three lambda model
This model showed the accuracy of survival description for higher or repeated doses.[7]
The equation used for this model is as follows:
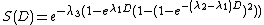
The linear-quadratic-cubic model
The equation used for this model is as follows:[7]
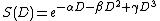
Sublesions hypothesis models
The repair-misrepair model
This model shows the mean number of lesions before any repair activations in a cell.[7]
The equation used for this model is as follows:
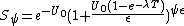
where Uo represents the yield of initially induced lesions, with λ being the linear self-repair coefficient, and T equaling time
The lethal-potentially lethal model
This equation explores the hypothesis of a lesion becoming fatal within a given of time if it is not repair by repair enzymes.[7]
The equation used for this model is as follows:
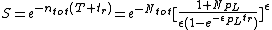
T is the radiation duration and tr is the available repair time.
The saturable repair model
This model illustrates the efficiency of the repair system decreasing as the dosage of radiation increases. This is due to the repair kinetics becoming increasingly saturated with the increase in radiation dosage.[7]
The equation used for this model is as follows:
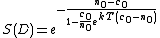
n(t) is the number of unrepaired lesions, c(t) is the number of repair molecules or enzymes, k is the proportionality coefficient, and T is the time available for repair.
Cellular environment and radiation hormesis
Radiation hormesis
Hormesis is the hypothesis that low levels of disrupting stimulus can cause beneficial adaptations in an organism.[8] The ionizing radiation stimulates repair proteins that are usually not active. Cells use this new stimuli to adapt to the stressors they are being exposed to.[9]Radiation-Induced Bystander Effect (RIBE)
In biology, the bystander effect is described as changes to nearby non-targeted cells in response to changes in an initially targeted cell by some disrupting agent.[10] In the case of Radiation-Induced Bystander Effect, the stress on the cell is caused by ionizing radiation.
The bystander effect can be broken down into two categories, long range bystander effect and short range bystander effect. In long range bystander effect, the effects of stress are seen further away from the initially targeted cell. In short range bystander, the effects of stress are seen in cells adjacent to the target cell.[10]
Both low linear energy transfer and high linear energy transfer photons have been shown to produce RIBE. Low linear energy transfer photons were reported to cause increases in mutagenesis and a reduction in the survival of cells in clonogenic assays. X-rays and gamma rays were reported to cause increases in DNA double strand break, methylation, and apoptosis.[10] Further studies are needed to reach a conclusive explanation of any epigenetic impact of the bystander effect.
Radiation and oxidative stress
Formation of ROS
Ionizing radiation produces fast moving particles which have the ability to damage DNA, and produce highly reactive free radicals known as reactive oxygen species (ROS). The production of ROS in cells radiated by LDIR (Low-Dose Ionizing Radiation) occur in two ways, by the radiolysis of water molecules or the promotion of nitric oxide synthesis (NOS) activity. The resulting nitric oxide formation reacts with superoxide radicals. This generates peroxynitrite which is toxic to biomolecules. Cellular ROS is also produced with the help of a mechanism involving nicotinamide adenosine dinucleotide phosphate (NADPH) oxidase. NADPH oxidase helps with the formation of ROS by generating a superoxide anion by transferring electrons from cytosolic NADPH across the cell membrane to the extracellular molecular oxygen. This process increases the potential for leakage of electrons and free radicals from the mitochondria. The exposure to the LDIR induces electron release from the mitochondria resulting in more electrons contributing to the superoxide formation in the cells.
The production of ROS in high quantity in cells results in the degradation of biomolecules such as proteins, DNA, and RNA. In one such instance the ROS are known to create double stranded and single stranded breaks in the DNA. This causes the DNA repair mechanisms to try to adapt to the increase in DNA strand breaks. Heritable changes to the DNA sequence have been seen although the DNA nucleotide sequence seems the same after the exposure with LDIR.[25]
Activation of NOS
The formation of ROS is coupled with the formation of nitric oxide synthase activity (NOS). NO reacts with O2− generating peroxynitrite. The increase in the NOS activity causes the production of peroxynitrite (ONOO-). Peroxynitrite is a strong oxidant radical and it reacts with a wide array of biomolecules such as DNA bases, proteins and lipids. Peroxynitrite affects biomolecules function and structure and therefore effectively destabilizes the cell.[25]
Mechanism of oxidative stress and epigenetic gene regulation
Ionizing radiation causes the cell to generate increased ROS and the increase of this species damages biological macromolecules. In order to compensate for this increased radical species, cells adapt to IR induced oxidative effects by modifying the mechanisms of epigenetic gene regulation. There are 4 epigenetic modifications that can take place:
- formation of protein adducts inhibiting epigenetic regulation
- alteration of genomic DNA methylation status
- modification of post translational histone interactions affecting chromatin compaction
- modulation of signaling pathways that control transcription factor expression
ROS-mediated protein adduct formation
ROS generated by ionizing radiation chemically modify histones which can cause a change in transcription. Oxidation of cellular lipid components result in an electrophilic molecule formation. The electrophilic molecule binds to the lysine residues of histones causing a ketoamide adduct formation. The ketoamide adduct formation blocks the lysine residues of histones from binding to acetylation proteins thus decreasing gene transcription.[27]
ROS-mediated DNA methylation changes
DNA hypermethylation is seen in the genome with DNA breaks at a gene-specific basis, such as promoters of regulatory genes, but the global methylation of the genome shows a hypomethylation pattern during the period of reactive oxygen species stress.[11]
DNA damage induced by reactive oxygen species results in increased gene methylation and ultimately gene silencing. Reactive oxygen species modify the mechanism of epigenetic methylation by inducing DNA breaks which are later repaired and then methylated by DNMTs. DNA damage response genes, such as GADD45A, recruit nuclear proteins Np95 to direct histone methyltransferase's towards the damaged DNA site. The breaks in DNA caused by the ionizing radiation then recruit the DNMTs in order to repair and further methylate the repair site.
Genome wide hypomethylation occurs due to reactive oxygen species hydroxylating methylcytosines to 5-hydroxymethylcytosine (5hmC).[12] The production of 5hmC serves as an epigenetic marker for DNA damage which is recognizable by DNA repair enzymes. The DNA repair enzymes attracted by the marker convert 5hmC to an unmethylated cytosine base resulting in the hypomethylation of the genome.[13]
Another mechanism that induces hypomethylation is the depletion of S-adenosyl methionine synthetase (SAM). The prevalence of super oxide species causes the oxidization of reduced glutathione (GSH) to GSSG. Due to this, synthesis of the cosubstrate SAM is stopped. SAM is an essential cosubtrate for the normal functioning of DNMTs and histone methyltrasnferase proteins.
ROS-mediated post-translation modification
Double stranded DNA breaks caused by exposure to ionizing radiation are known to alter chromatin structure. Double stranded breaks are primarily repaired by poly ADP (PAR) polymerases which accumulate at the site of the break leading to activation of the chromatin remodeling protein ALC1. ALC1 causes the nucleosome to relax resulting in the epigenetic up-regulation of genes. A similar mechanism involves the ataxia telangiectasia mutated (ATM) serine/threonine kinase which is an enzyme involved in the repair of double stranded breaks caused by ionizing radiation. ATM phosphorylates KAP1 which causes the heterochromatin to relax, allowing increased transcription to occur.
The DNA mismatch repair gene (MSH2) promoter has shown a hypermethylation pattern when exposed to ionizing radiation. Reactive oxygen species induce the oxidization of deoxyguanosine into 8-hydroxydeoxyguanosine (8-OHdG) causing a change in chromatin structure. Gene promoters that contain 8-OHdG deactivate the chromatin by inducing trimethyl-H3K27 in the genome. Other enzymes such as transglutaminases (TGs) control chromatin remodeling through proteins such as sirtuin1 (SIRT1). TGs cause transcriptional repression during reactive oxygen species stress by binding to the chromatin and inhibiting the sirtuin 1 histone deacetylase from performing its function.[27]
ROS-mediated loss of epigenetic imprinting
Epigenetic imprinting is lost during reactive oxygen species stress. This type of oxidative stress causes a loss of NF- κB signaling. Enhancer blocking element CCCTC-binding factor (CTCF) binds to the imprint control region of insulin-like growth factor 2 (IGF2) preventing the enhancers from allowing the transcription of the gene. The NF- κB proteins interact with IκB inhibitory proteins, but during oxidative stress IκB proteins are degraded in the cell. The loss of IκB proteins for NF- κB proteins to bind to results in NF- κB proteins entering the nucleus to bind to specific response elements to counter the oxidative stress. The binding of NF- κB and corepressor HDAC1 to response elements such as the CCCTC-binding factor causes a decrease in expression of the enhancer blocking element. This decrease in expression hinders the binding to the IGF2 imprint control region therefore causing the loss of imprinting and biallelic IGF2 expression.[27]Mechanisms of epigenetic modifications
After the initial exposure to ionizing radiation, cellular changes are prevalent in the unexposed offspring of irradiated cells for many cell divisions. One way this non-Mendelian mode of inheritance can be explained is through epigenetic mechanisms.[14]
Ionizing radiation and DNA methylation
Genomic instability via hypomethylation of LINE1
Ionizing radiation exposure affects patterns of DNA methylation. Breast cancer cells treated with fractionated doses of ionizing radiation showed DNA hypomethylation at the various gene loci; dose fractionation refers to breaking down one dose of radiation into separate, smaller doses.[15] Hypomethylation of these genes correlated with decreased expression of various DNMTs and methyl CpG binding proteins. LINE1 transposable elements have been identified as targets for ionizing radiation. The hypomethylation of LINE1 elements results in activation of the elements and thus an increase in LINE1 protein levels. Increased transcription of LINE1 transposable elements results in greater mobilization of the LINE1 loci and therefore increases genomic instability.[16]
Ionizing radiation and histone modification
Irradiated cells can be linked to a variety of histone modifications. Ionizing radiation in breast cancer cell inhibits H4 lysine tri-methylation. Mouse models exposed to high levels of X-ray irradiation exhibited a decrease in both the tri-methylation of H4-Lys20 and the compaction of the chromatin. With the loss of tri-methylation of H4-Lys20, DNA hypomethylation increased resulting in DNA damage and increased genomic instability.[14]
Loss of methylation via repair mechanisms
Breaks in DNA due to ionizing radiation can be repaired. New DNA synthesis by DNA polymerases is one of the ways radiation induced DNA damage can be repaired. However, DNA polymerases do not insert methylated bases which leads to a decrease in methylation of the newly synthesized strand. Reactive oxygen species also inhibit DNMT activity which would normally add the missing methyl groups. This increases the chance that the demethylated state of DNA will eventually become permanent.[17]
Clinical consequences and applications
Epigenetic affects on a developing brain
Chronic exposure to these types of radiation can have an effect on children from as early as when they are fetuses. There have been multiple cases reported of hindrance in the development of the brain, behavioral changes such as anxiety, and the disruption of proper learning and language processing. An Increase in the cases of ADHD behavior and autism behavior has been shown to be directly correlated with the exposure of EMF waves. The World Health Organization has classified RFR as a possible carcinogen for its epigenetic effects on DNA expression. The exposure to EMF waves on a consistent 24hr basis has shown to lower the activity of miRNA in the brain affecting developmental and neuronal activity. This epigenetic change causes the silencing of necessary genes along with the change in expression of other genes integral for the normal development of the brain.[2]
MGMT- and LINE1- specific DNA methylation
DNA methylation influences tissue responses to ionizing radiation. Modulation of methylation in the gene MGMT or in transposable elements such as LINE1 could be used to alter tissue responses to ionizing radiation and potentially opening new areas for cancer treatment.
MGMT serves as a prognostic marker in glioblastoma. Hypermethylation of MGMT is associated with the regression of tumors. Hypermethylation of MGMT silences its transcription inhibiting alkylating agents in tumor killing cells. Studies have shown patients who received radiotherapy, but no chemotherapy after tumor extraction, had an improved response to radiotherapy due to the methylation of the MGMT promoter.
Almost all human cancers include hypomethylation of LINE1 elements. Various studies depict that the hypomethylation of LINE1 correlates with a decrease in survival after both chemotherapy and radiotheraphy.
Treatment by DNMT inhibitors
DMNT inhibitors are being explored in the treatment of malignant tumors. Recent in-vitro studies show that DNMT inhibitors can increase the effects of other anti-cancer drugs. Knowledge of in-vivo effect of DNMT inhibitors are still being investigated. The long term effects of the use of DNMT inhibitors are still unknown.[17]References
2. ^1 2 {{Cite journal|last=Sage|first=Cindy|last2=Burgio|first2=Ernesto|date=2018-01-01|title=Electromagnetic Fields, Pulsed Radiofrequency Radiation, and Epigenetics: How Wireless Technologies May Affect Childhood Development|journal=Child Development|language=en|volume=89|issue=1|pages=129–136|doi=10.1111/cdev.12824|pmid=28504324|issn=1467-8624}}
3. ^{{Cite journal|last=de Siqueira|first=Elisa Carvalho|last2=de Souza|first2=Fabrício Tinoco Alvim|last3=Gomez|first3=Ricardo Santiago|last4=Gomes|first4=Carolina Cavalieri|last5=de Souza|first5=Renan Pedra|date=2017-01-24|title=Does cell phone use increase the chances of parotid gland tumor development? A systematic review and meta-analysis|journal=Journal of Oral Pathology & Medicine|volume=46|issue=7|pages=480–483|doi=10.1111/jop.12531|pmid=27935126|issn=0904-2512}}
4. ^1 {{Cite web|url=http://jcdr.net/article_fulltext.asp?issn=0973-709x&year=2017&volume=11&issue=5&page=ZE01&issn=0973-709x&id=9883|title=JCDR - Non ionizing radiations, Oral cavity, Salivary gland, Tumour|website=jcdr.net|language=en|access-date=2018-05-10}}
5. ^{{cite book|last1=IARC|title=Non-Ionizing Radiation, Part 2: Radiofrequency Electromagnetic Fields|date=2013|publisher=World Health Organisation|isbn=978 92 832 1325 3|url=http://monographs.iarc.fr/ENG/Monographs/vol102/index.php|volume=102|series=IARC Monographs on the Evaluation of Carcinogenic Risks to Humans}}
6. ^{{Cite journal|last=Summers|first=William C|date=|title=Physics and Genes: Einstein to Delbrück|url=|journal=Creating a Physical Biology:The Three-Man Paper and Early Molecular Biology|volume=|pages=39–68|via=}}
7. ^1 2 3 4 5 6 7 8 {{Cite journal|last=Bodgi|first=Larry|last2=Canet|first2=Aurelien|last3=Pujo-Menjouet|first3=Laurent|last4=Lesne|first4=Annick|last5=Victor|first5=Jean-Marc|last6=Foray|first6=Nicolas|date=2016-04-07|title=Mathematical models of radiation action on living cells: From the target theory to the modern approaches. A historical and critical review|journal=Journal of Theoretical Biology|language=en|volume=394|pages=93–101|doi=10.1016/j.jtbi.2016.01.018|pmid=26807808|issn=0022-5193}}
8. ^{{Cite journal|last=Vaiserman|first=Alexander M.|accessdate=April 1, 2018|year=2011|title=Hormesis and epigenetics: Is there a link?|url=https://ac-els-cdn-com.ezproxy.lib.utexas.edu/S1568163711000055/1-s2.0-S1568163711000055-main.pdf?_tid=28b1af2b-89f9-4efd-8833-c8280bbd8ea6&acdnat=1522641757_9cde4ff1b14f4fd87539791c6d41bcc9|journal=Ageing Research Reviews|publication-date=September 2011|volume=10|issue=4|pages=413–421|via=Web of Science}}
9. ^{{Cite journal|last=Kim|first=Se-A.|last2=Lee|first2=Yu-Mi|last3=Choi|first3=Je-Yong|last4=Jacobs|first4=David R.|last5=Lee|first5=Duk-Hee|title=Evolutionarily adapted hormesis-inducing stressors can be a practical solution to mitigate harmful effects of chronic exposure to low dose chemical mixtures|journal=Environmental Pollution|volume=233|pages=725–734|doi=10.1016/j.envpol.2017.10.124|pmid=29126094|year=2018}}
10. ^1 2 {{Cite journal|last=Verma|first=Neha|last2=Tiku|first2=Ashu Bhan|access-date=Jun 27, 2018|title=Significance and nature of bystander responses induced by various agents|url=https://www.sciencedirect.com/science/article/pii/S138357421630014X|journal=Mutation Research/Reviews in Mutation Research|date=Jul 2017|volume=773|pages=104–121|via=Web of Science|doi=10.1016/j.mrrev.2017.05.003|pmid=28927522}}
11. ^{{Cite journal|date=2017-10-23|title=Epigenetics of malignant melanoma|journal=Seminars in Cancer Biology|volume=51|pages=80–88|language=en|doi=10.1016/j.semcancer.2017.10.006|pmid=29074395|issn=1044-579X|last1=Moran|first1=Bruce|last2=Silva|first2=Romina|last3=Perry|first3=Antoinette S.|last4=Gallagher|first4=William M.}}
12. ^{{Cite journal|last=Ni|first=Qihan Wu and Xiaohua|date=2014-12-31|title=ROS-Mediated DNA Methylation Pattern Alterations in Carcinogenesis|url=http://www.eurekaselect.com/127591/article|journal=Current Drug Targets|language=en|volume=16|issue=1}}
13. ^{{Cite journal|last=Kafer|first=Georgia Rose|last2=Li|first2=Xuan|last3=Horii|first3=Takuro|last4=Suetake|first4=Isao|last5=Tajima|first5=Shoji|last6=Hatada|first6=Izuho|last7=Carlton|first7=Peter Mark|date=Feb 4, 2016|title=5-Hydroxymethylcytosine Marks Sites of DNA Damage and Promotes Genome Stability|url=https://www.cell.com/cell-reports/fulltext/S2211-1247(16)00056-5?_returnURL=https://linkinghub.elsevier.com/retrieve/pii/S2211124716000565?showall=true|journal=Cell Reports|language=English|volume=14|issue=6|pages=1283–1292|doi=10.1016/j.celrep.2016.01.035|pmid=26854228|issn=2211-1247}}
14. ^1 2 3 4 {{Cite journal|last=Tharmalingam|first=Sujeenthar|last2=Sreetharan|first2=Shayenthiran|last3=Kulesza|first3=Adomas V.|last4=Boreham|first4=Douglas R.|last5=Tai|first5=T. C.|date=Jul 28, 2017|title=Low-Dose Ionizing Radiation Exposure, Oxidative Stress and Epigenetic Programing of Health and Disease|journal=Radiation Research|volume=188|issue=4.2|pages=525–538|doi=10.1667/RR14587.1|pmid=28753061|issn=0033-7587|bibcode=2017RadR..188..525T}}
15. ^{{Cite web|url=https://www.cancer.gov/publications/dictionaries/cancer-terms|title=NCI Dictionary of Cancer Terms|website=National Cancer Institute|language=en|access-date=2018-05-10|date=2011-02-02}}
16. ^1 2 {{Cite journal|last=Tharmalingam|first=Sujeenthar|last2=Sreetharan|first2=Shayenthiran|last3=Kulesza|first3=Adomas V.|last4=Boreham|first4=Douglas R.|last5=Tai|first5=T. C.|date=Jul 28, 2017|title=Low-Dose Ionizing Radiation Exposure, Oxidative Stress and Epigenetic Programing of Health and Disease|journal=Radiation Research|volume=188|issue=4.2|pages=525–538|doi=10.1667/rr14587.1|pmid=28753061|bibcode=2017RadR..188..525T}}
17. ^1 {{Cite journal|last=Miousse|first=Isabelle R.|last2=Kutanzi|first2=Kristy R.|last3=Koturbash|first3=Igor|date=Feb 21, 2017|title=Effects of Ionizing Radiation on DNA Methylation: From Experimental Biology to Clinical Applications|journal=International Journal of Radiation Biology|volume=93|issue=5|pages=457–469|doi=10.1080/09553002.2017.1287454|issn=0955-3002|pmc=5411327|pmid=28134023}}
- Back from the Grave, Volume 1 (LP)
- Back from the Grave, Volume 2
- Back from the Grave, Volume 2 (CD)
- Back from the Grave, Volume 2 (LP)
- Back from the Grave, Volume 3
- Back from the Grave, Volume 3 (CD)
- Back from the Grave, Volume 3 (LP)
- Back from the Grave, Volume 4
- Back from the Grave, Volume 4 (CD)
- Back from the Grave, Volume 4 (LP)
- Back from the Grave, Volume 5
- Back from the Grave, Volume 5 (LP)
- Back from the Grave, Volume 6
- Back from the Grave, Volume 6 (LP)
- Back from the Grave, Volume 7
- Back from the Grave, Volume 8 (CD)
- Back from the Grave, Volume 8 (LP)
- Back from the Grave, Volume 9
- Back from the Grave, Volume 9 (LP)
- Back from the Rains
- Back from the Sewers
- Back From the Shadows: The Best of the Firesign Theatre
- BACKG (DOS command)
- Back Gill
- Back Glass