- Background
- Rigorous Formulation Determination of VB Structures Rumer's Method Matrix Representation of VB Structures Definition of Chirgwin Coulson Weights Half Determinant Decomposition of Molecular Orbitals
- Sample Computations for Simple Molecules Computations for the Hydrogen Molecule Computations for Ozone
- Applications to Main Group Compounds Borazine S2N2
- References
In modern valence bond (VB) theory calculations, Chirgwin–Coulson weights (also called Mulliken weights) are the relative weights of a set of possible VB structures of a molecule. Related methods of finding the relative weights of valence bond structures are the Löwdin[1] and the inverse weights.[2]
Background
For a wave function 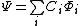 where
where 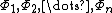 are a linearly independent, orthogonal set of basis orbitals, the weight of a constituent orbital
are a linearly independent, orthogonal set of basis orbitals, the weight of a constituent orbital 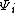 would be
would be 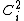 since the overlap integral,
since the overlap integral,  , between two wave functions
, between two wave functions 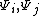 would be 1 for
would be 1 for 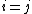 and 0 for
and 0 for  . In valence bond theory, however, the generated structures are not necessarily orthogonal with each other, and often times have substantial overlap between the two structures. As such, when considering non-orthogonal constituent orbitals (i.e. orbitals with non-zero overlap) the non-diagonal terms in the overlap matrix would be non-zero, and must be included in determining the weight of a constituent orbital. A method of computing the weight of a constituent orbital,
. In valence bond theory, however, the generated structures are not necessarily orthogonal with each other, and often times have substantial overlap between the two structures. As such, when considering non-orthogonal constituent orbitals (i.e. orbitals with non-zero overlap) the non-diagonal terms in the overlap matrix would be non-zero, and must be included in determining the weight of a constituent orbital. A method of computing the weight of a constituent orbital,  , proposed by Chirgwin and Coulson would be:[2]
, proposed by Chirgwin and Coulson would be:[2]
 }}
}}Application of the Chirgwin-Coulson formula to a molecular orbital yields the Mulliken population of the molecular orbital.[3]
Rigorous Formulation
Determination of VB Structures
Rumer's Method
A method of creating a linearly independent, complete set of valence bond structures for a molecule was proposed by Yuri Rumer. For a system with n electrons and n orbitals, Rumer's method involves arranging the orbitals in a circle and connecting the orbitals together with lines that do not intersect one another.[4] Covalent, or uncharged, structures can be created by connecting all of the orbitals with one another. Ionic, or charged, structures for a given atom can be determined by assigning a charge to a molecule, and then following Rumer's method. For the case of butadiene, the 20 possible Rumer structures are shown, where 1 and 2 are the covalent structures, 3-14 are the monoionic structures, and 15-20 are the diionic structures. The resulting VB structures can be represented by a linear combination of determinants 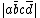 , where a letter without an over-line indicates an electron with
, where a letter without an over-line indicates an electron with  spin, while a letter with over-line indicates an electron with
spin, while a letter with over-line indicates an electron with 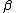 spin. The VB structure for 1, for example would be a linear combination of the determinants
spin. The VB structure for 1, for example would be a linear combination of the determinants 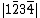 ,
, 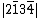 ,
,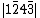 , and
, and 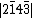 . For a monoanionic species, the VB structure for 11 would be a linear combination of
. For a monoanionic species, the VB structure for 11 would be a linear combination of 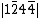 and
and 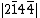 , namely:
, namely:
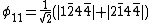
Matrix Representation of VB Structures
An arbitrary VB structure 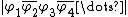 containing
containing  electrons, represented by the electron indices
electrons, represented by the electron indices 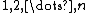 , and orbitals, represented by
, and orbitals, represented by 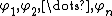 , can be represented by the following Slater determinant:
, can be represented by the following Slater determinant:
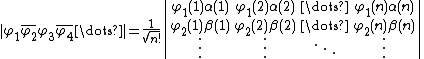
Where  and
and 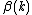 represent an or spin on the
represent an or spin on the 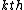 electron, respectively. For the case of a two electron system with orbitals
electron, respectively. For the case of a two electron system with orbitals  and
and  , the VB structure,
, the VB structure, 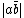 , can be represented:
, can be represented: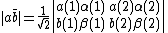
Evaluating the determinant yields:[5]
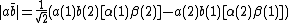
Definition of Chirgwin Coulson Weights
Given a wave function where 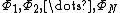 is a complete, linearly independent set of VB structures and
is a complete, linearly independent set of VB structures and  is the coefficient of each structure, the Chirgwin-Coulson weight
is the coefficient of each structure, the Chirgwin-Coulson weight 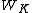 of a VB structure
of a VB structure 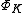 can be computed in the following manner:[2]
can be computed in the following manner:[2]
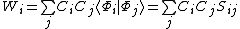
Where  is the overlap matrix satisfying
is the overlap matrix satisfying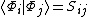 .
.
Other methods of computing weights of VB structure include Löwdin weights, where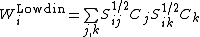 ,[1] and inverse weights, where
,[1] and inverse weights, where 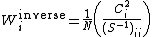 with
with 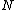 being a normalization factor defined by
being a normalization factor defined by 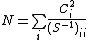 .[6] The use of Löwdin and inverse weights is appropriate when the Chirgwin–Coulson weights either exceed 1 or are negative.[6]
.[6] The use of Löwdin and inverse weights is appropriate when the Chirgwin–Coulson weights either exceed 1 or are negative.[6]
Half Determinant Decomposition of Molecular Orbitals
Given a set of molecular orbitals, 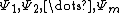 , for a molecule, consider the determinant of a given orbital population, represented by
, for a molecule, consider the determinant of a given orbital population, represented by 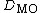 . The determinant can be written as the following Slater determinant:
. The determinant can be written as the following Slater determinant:
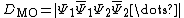
Computing the determinant explicitly by multiplying this expression can be a computationally difficult task, given that each molecular orbital is composed of a combination of atomic orbitals. On the other hand, because the determinant of a product of matrices is equal to the product of determinants, the determinant can be regrouped to half-determinants, one of which contains only electrons with spin and the only with electrons of spin, that is: 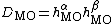 where
where  and
and 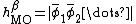 .[5][7][13]
.[5][7][13]
Note that any given molecular orbital 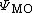 can be written as a linear combination of atomic orbitals
can be written as a linear combination of atomic orbitals 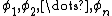 , that is for each , there exist
, that is for each , there exist  such that
such that 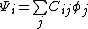 . As such, the half determinant
. As such, the half determinant 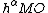 can be further decomposed into the half determinants for an ordering of atomic orbitals
can be further decomposed into the half determinants for an ordering of atomic orbitals 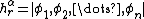 corresponding to a VB structure
corresponding to a VB structure  . As such, the molecular orbital can be represented as a combination of the half determinants of the atomic orbitals,
. As such, the molecular orbital can be represented as a combination of the half determinants of the atomic orbitals, 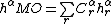 . The coefficient
. The coefficient 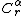 can be determined by evaluating the following matrix:[5][7][13]
can be determined by evaluating the following matrix:[5][7][13]
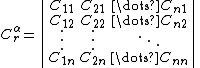
The same method can be used to evaluate the half determinant for the electrons, 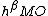 . As such, the determinant
. As such, the determinant 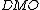 can be expressed as
can be expressed as 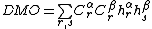 , where
, where 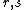 index across all possible VB structures.[5][7][13]
index across all possible VB structures.[5][7][13]
Sample Computations for Simple Molecules
Computations for the Hydrogen Molecule
The hydrogen molecule can be considered to be a linear combination of two  orbitals, indicated as
orbitals, indicated as  and
and  . The possible VB structures for
. The possible VB structures for 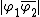 and
and  indicated as 1 and 2 respectively, as well as the ionic structures
indicated as 1 and 2 respectively, as well as the ionic structures 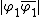 and
and 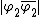 indicated as 3 and 4 respectively, shown below.
indicated as 3 and 4 respectively, shown below.
Because structures 1 and 2 both represent covalent bonding in the hydrogen molecule and exchanging the electrons of structure 1 yields structure 2, the two covalent structures can be combined into one wave function. As such, the Heitler-London model for bonding in 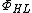 , can be used in place of the VB structures and
, can be used in place of the VB structures and  :[8]
:[8]
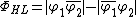
Where the negative sign arises from the antisymmetry of electron exchange. As such, the wave function for the 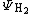 , can be considered to be a linear combination of the Heitler-London structure and the two ionic valence bond structures.
, can be considered to be a linear combination of the Heitler-London structure and the two ionic valence bond structures.
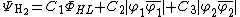
The overlap matrix between the atomic orbitals between the three valence bond configurations , , and is given in the output for valence bond calculations. A sample output is given below:[5]
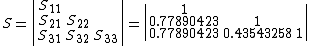
Finding the eigenvectors of the matrix 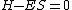 , where
, where  is the hamiltonian and
is the hamiltonian and  is energy due to orbital overlap, yields the VB-vector
is energy due to orbital overlap, yields the VB-vector  , which satisfies:[9]
, which satisfies:[9]
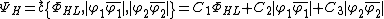
Solving for the VB-vector using density functional theory yields the coefficients 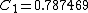 and
and 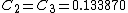 . Thus, the Coulson-Chrigwin weights can be computed:[5]
. Thus, the Coulson-Chrigwin weights can be computed:[5]
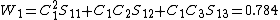

To check for consistency, the inverse weights can be computed by first determining the inverse of the overlap matrix:
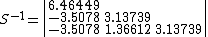
Next, the normalization constant  can be determined:
can be determined:
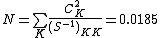
The final weights are: 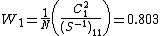 , and
, and 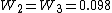 .
.
Informally, the computed weights indicate that the wave function for the
Computations for Ozone
Determining the relative weights of each resonance structure of ozone requires, first, the determination of the possible VB structures for  orbitals of oxygen, and labeling the orbital on the
orbitals of oxygen, and labeling the orbital on the  oxygen
oxygen  ,
,  structure can be represented by the determinants
structure can be represented by the determinants 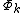 :[5]
:[5]
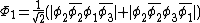
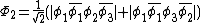
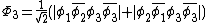
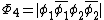
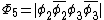
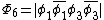
orbitals are in phase, one where there is a node on the central oxygen, and one where all of the oxygen orbitals are out of phase, shown below:
The wave functions for each of the molecular orbitals  can be written as a linear combination of each of the oxygen orbitals as follows:[5]
can be written as a linear combination of each of the oxygen orbitals as follows:[5]
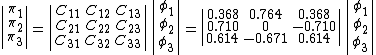
Where indicates the coefficient of  in a molecular orbital . Consider, the VB contributions for the ground state of
in a molecular orbital . Consider, the VB contributions for the ground state of 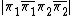 . Using the methods of half determinants, the half determinants for the ground state are:
. Using the methods of half determinants, the half determinants for the ground state are:

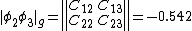
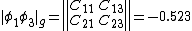
By the method of half determinant expansion, the coefficient, 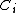 , for a structure
, for a structure 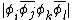 is:
is:
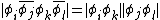
Which implies that the ground state has the following coefficients:
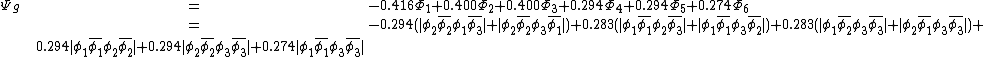
Given the following overlap matrix for the half determinants:[5]
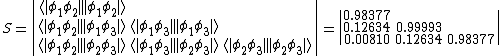
The overlap between two VB structures represented by the product of two VB determinants 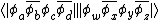 can be evaluated by finding the product of the overlap between the two half determinants, that is:
can be evaluated by finding the product of the overlap between the two half determinants, that is:

For example, the overlap between the orbitals 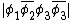 and
and 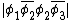 would be:
would be:

The weights of the standard Lewis structures for 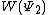 and
and 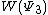 . The weights can be found by first computing the Chirgwin–Coulson weights for their constituent determinants:
. The weights can be found by first computing the Chirgwin–Coulson weights for their constituent determinants:
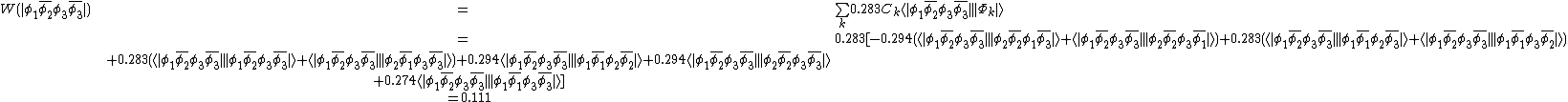
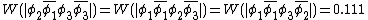
The weights for the standard lewis structures would be the sum of the weights of the constituent determinants. As such:[2]
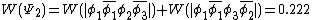
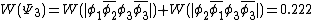
This compares well with reported Chirgwin–Coulson weights of 0.226 for the standard Lewis structure of ozone in the ground state.[10]
For the diradical state, 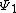 , the weight is:
, the weight is:
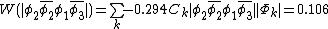
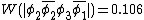
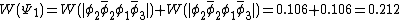
This also compares favorably with reported Chirgwin–Coulson weights of 0.213 for the diradical state of ozone in the ground state.[10]
Applications to Main Group Compounds
Borazine
Borazine, (chemical formula
However, VB calculations using a double‐zeta D95 basis set indicate that the predominant resonance structures are the structure with all three lone pairs on the nitrogen (labeled 1 below) and the six resonance structures with one double bond between boron and nitrogen (labeled 2 below). The relative weights of the two structures are 0.17 and 0.08 respectively.[11][12]
By contrast, the dominant resonance structures of benzene are the two Kekule structures, with weight 0.15, and 12 monozwitterionic structures with weight 0.03. The data, together, indicate that, despite the similarity in appearance and structure, the electrons on borazine are less delocalized than those on benzene.[11]
S2N2
Disulfur dinitride is a square planar compound that contains a 6 electron conjugated  system. The primary diradical resonance structures (1 and 2) and a secondary zwitterionic structure (3) are shown below:
system. The primary diradical resonance structures (1 and 2) and a secondary zwitterionic structure (3) are shown below:
Valence bond calculations using the Dunning's D95 full double-zeta basis set indicate that the dominant resonance structure is the singlet diradical with a long nitrogen-nitrogen bond (structure 1), with Chirgwin-Coulson weight 0.47. This value is substantially higher than the weight for the singlet diradical centered on the sulfurs (structure 2), which has a Chirgwin-Coulson weight of 0.06.[13] This result corresponds nicely with the general rules regarding Lewis structures, namely that formal charges ought to be minimized, and contrasts with earlier computational results indicating that 1 is the dominant structure.[14]
References
2. ^1 2 {{Cite journal|last=Chirgwin|first=B. H.|last2=Coulson|first2=C. A.|date=22 March 1950|title=The Electronic Structure of Conjugated Systems. VI |journal=Proceedings of the Royal Society A: Mathematical, Physical and Engineering Sciences|volume=201|issue=1065|pages=196–209|doi=10.1098/rspa.1950.0053|issn=1364-5021|bibcode=1950RSPSA.201..196C}}
3. ^{{Cite journal|last=Mulliken|first=R. S.|date=October 1955 |title=Electronic Population Analysis on LCAO–MO Molecular Wave Functions. I |journal=The Journal of Chemical Physics|volume=23|issue=10|pages=1833–1840|doi=10.1063/1.1740588|issn=0021-9606|bibcode=1955JChPh..23.1833M}}
4. ^{{Cite journal|last=Rumer|first=Georg|date=1932|title=Zur Theorie der Spinvalenz |url=https://gdz.sub.uni-goettingen.de/id/PPN252457811_1932?tify={%22pages%22:[341],%22panX%22:0.569,%22panY%22:0.825,%22view%22:%22info%22,%22zoom%22:0.52}|journal=Gottinger Nachr.|volume=|pages=337}}
5. ^1 2 3 4 5 6 7 8 {{Cite book|doi=10.1002/9780470192597 |title=A Chemist's Guide to Valence Bond Theory|last=Shaik|first=Sason|last2=Hiberty|first2=Philippe C.|date=2007-11-16|publisher=John Wiley & Sons, Inc.|isbn=9780470192597|location=Hoboken, NJ, USA}}
6. ^1 2 {{Cite journal|last=Gallup|first=G.A.|last2=Norbeck|first2=J.M.|date=September 1973 |title=Population analyses of valence-bond wavefunctions and BeH2 |journal=Chemical Physics Letters|volume=21|issue=3|pages=495–500|doi=10.1016/0009-2614(73)80292-1|issn=0009-2614|bibcode=1973CPL....21..495G}}
7. ^1 2 {{Cite book|last=Shaik|first=Sason|chapter=Valence Bond Theory, Its History, Fundamentals, and Applications: A Primer|date=2004-08-17 |doi=10.1002/0471678856.ch1|title=Reviews in Computational Chemistry|pages=1–100|publisher=John Wiley & Sons, Inc.|isbn=9780471445258 |last2=Hiberty|first2=Philippe C.}}
8. ^{{Cite journal|last=Heitler|first=W.|last2=London|first2=F.|date=June 1927 |title=Wechselwirkung neutraler Atome und homöopolare Bindung nach der Quantenmechanik|journal=Zeitschrift für Physik|volume=44|issue=6–7|pages=455–472|doi=10.1007/bf01397394|issn=1434-6001|bibcode=1927ZPhy...44..455H}}
9. ^J. Verbeek, J. H. Langenberg, C. P. Byrman, F. Dijkstra, J. H. van Lenthe, TURTLE: An Ab Initio VB/VBSCF Program (1998–2000)
10. ^1 2 3 4 {{Cite journal|last=Hiberty|first=P. C.|last2=Leforestier|first2=C.|date=March 1978|title=Expansion of molecular orbital wave functions into valence bond wave functions. A simplified procedure |journal=Journal of the American Chemical Society|volume=100|issue=7|pages=2012–2017|doi=10.1021/ja00475a007|issn=0002-7863}}
11. ^1 {{Cite journal|last=Benker|first=Daniel|last2=Klapötke|first2=Thomas M.|last3=Kuhn|first3=Gerhard|last4=Li|first4=Jiabo|last5=Miller|first5=Christian|date=2005|title=An ab initio valence bond (VB) calculation of the π delocalization energy in borazine, B3N3H6|journal=Heteroatom Chemistry|volume=16|issue=5|pages=311–315|doi=10.1002/hc.20095|issn=1042-7163}}
12. ^Engelberts, Jeroen Johan. “Analysis Of Chemical Bonding Using Ab Initio Valence Bond Theory.” Utrecht University, 2017.
13. ^{{Cite journal|last=Klapoetke|first=Thomas M.|last2=Li|first2=Jiabo|last3=Harcourt|first3=Richard D.|date=2004-10-12|title=Ab initio Double-ζ (D95) Valence Bond Calculations for the Ground States of S2N2 and S42+ |journal=ChemInform|volume=35|issue=41|doi=10.1002/chin.200441002|issn=0931-7597}}
14. ^{{Cite journal|last=Gerratt|first=J.|last2=McNicholas|first2=S. J.|last3=Karadakov|first3=P. B. |last4=Sironi|first4=M.|last5=Raimondi|first5=M.|last6=Cooper|first6=D. L.|date=January 1996|title=The Extraordinary Electronic Structure of N2S2 |journal=Journal of the American Chemical Society|volume=118|issue=27|pages=6472–6476|doi=10.1021/ja953994f|issn=0002-7863}}
- Consten SaRL and Grundig GmbH v Commission
- Constilac
- Constimol
- Constitucion del Estado de Nuevo Mexico
- Constitucion District
- Constitucion (Line C Buenos Aires Metro)
- Constitucion Unido
- Constitucion, Uruguay
- Constitución, Baja California Sur
- Constitución-class battleship
- Constitución del Estado de Nuevo Mexico
- Constitución del Estado de Nuevo México
- Constitución District
- Constitución Española
- Constitución (Line C Buenos Aires Metro)
- Constitución (Line E Buenos Aires Underground)
- Constitución Unido
- Constitución, Uruguay
- Constituencies for French residents overseas
- Constituencies in Solomon Islands
- Constituencies of Jamaica
- Constituencies of Tanzania
- Constituency Development Fund
- Constituency (disambiguation)
- Constituency Labour parties