- Origin
- Method B3LYP PBE0 HSE Meta hybrid GGA
- References
Hybrid functionals are a class of approximations to the exchange–correlation energy functional in density functional theory (DFT) that incorporate a portion of exact exchange from Hartree–Fock theory with the rest of the exchange-correlation energy from other sources (ab initio or empirical). The exact exchange energy functional is expressed in terms of the Kohn–Sham orbitals rather than the density, so is termed an implicit density functional. One of the most commonly used versions is B3LYP, which stands for Becke, 3-parameter, Lee-Yang-Parr.
Origin
The hybrid approach to constructing density functional approximations was introduced by Axel Becke in 1993.[1] Hybridization with Hartree–Fock (exact) exchange provides a simple scheme for improving the calculation of many molecular properties, such as atomization energies, bond lengths and vibration frequencies, which tend to be poorly described with simple "ab initio" functionals.[2]
Method
A hybrid exchange-correlation functional is usually constructed as a linear combination of the Hartree–Fock exact exchange functional, 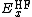 :
:
,
and any number of exchange and correlation explicit density functionals. The parameters determining the weight of each individual functional are typically specified by fitting the functional's predictions to experimental or accurately calculated thermochemical data, although in the case of the "adiabatic connection functionals" the weights can be set a priori.[2]
B3LYP
For example, the popular B3LYP (Becke, three-parameter, Lee-Yang-Parr)[3][4] exchange-correlation functional is:
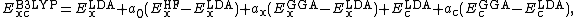
where 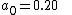 ,
,  , and
, and 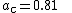 .
. 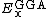 and
and 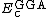 are generalized gradient approximations: the Becke 88 exchange functional[5] and the correlation functional of Lee, Yang and Parr[6] for B3LYP, and
are generalized gradient approximations: the Becke 88 exchange functional[5] and the correlation functional of Lee, Yang and Parr[6] for B3LYP, and 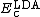 is the VWN local-density approximation to the correlation functional.[7]
is the VWN local-density approximation to the correlation functional.[7]
The three parameters defining B3LYP have been taken without modification from Becke's original fitting of the analogous B3PW91 functional to a set of atomization energies, ionization potentials, proton affinities, and total atomic energies.[8]
PBE0
The PBE0 functional[2]
[9]mixes the Perdew–Burke-Ernzerhof (PBE) exchange energy and Hartree-Fock exchange energy in a set 3 to 1 ratio, along with the full PBE correlation energy:
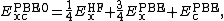
where 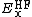 is the Hartree–Fock exact exchange functional,
is the Hartree–Fock exact exchange functional, 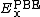 is the PBE exchange functional, and
is the PBE exchange functional, and 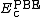 is the PBE correlation functional.[10]
is the PBE correlation functional.[10]
HSE
The HSE (Heyd-Scuseria-Ernzerhof)[11] exchange-correlation functional uses an error function screened Coulomb potential to calculate the exchange portion of the energy in order to improve computational efficiency, especially for metallic systems.
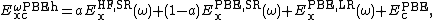
where  is the mixing parameter and
is the mixing parameter and 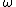 is an adjustable parameter controlling the short-rangeness of the interaction. Standard values of
is an adjustable parameter controlling the short-rangeness of the interaction. Standard values of 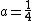 and
and 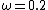 (usually referred to as HSE06) have been shown to give good results for most systems. The HSE exchange-correlation functional degenerates to the PBE0 hybrid functional for
(usually referred to as HSE06) have been shown to give good results for most systems. The HSE exchange-correlation functional degenerates to the PBE0 hybrid functional for  .
. 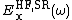 is the short range Hartree–Fock exact exchange functional,
is the short range Hartree–Fock exact exchange functional, 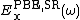 and
and 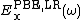 are the short and long range components of the PBE exchange functional, and
are the short and long range components of the PBE exchange functional, and 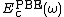 is the PBE [10] correlation functional.
is the PBE [10] correlation functional.
Meta hybrid GGA
The M06 suite of functionals[12][13] is a set of four meta-hybrid GGA and meta-GGA DFT functionals. These functionals are constructed by empirically fitting their parameters, while being constrained to a uniform electron gas.
The family includes the functionals M06-L, M06, M06-2X and M06-HF, with a different amount of exact exchange for each one. M06-L is fully local without HF exchange (thus it cannot be considered hybrid), M06 has 27% HF exchange, M06-2X 54% and M06-HF 100%.
The advantages and usefulness of each functional are
- M06-L: Fast, Good for transition metals, inorganic and organometallics.
- M06: For main group, organometallics, kinetics and non-covalent bonds.
- M06-2X: Main group, kinetics.
- M06-HF: Charge transfer TD-DFT, systems where self interaction is pathological.
The suite gives good results for systems containing dispersion forces, one of the biggest deficiencies of standard DFT methods. The s6 scaling factors on Grimme's long-range dispersion correction are 0.20, 0.25 and 0.06 for M06-L, M06 and M06-2X, respectively.
References
2. ^1 2 {{cite journal|author1=John P. Perdew |author2=Matthias Ernzerhof |author3=Kieron Burke |title=Rationale for mixing exact exchange with density functional approximations|journal=J. Chem. Phys.|volume=105|pages=9982–9985|year=1996|url=http://dft.uci.edu/pubs/PEB96.pdf|doi=10.1063/1.472933|accessdate=2007-05-07|bibcode = 1996JChPh.105.9982P|issue=22 }}
3. ^{{ cite journal |author1=K. Kim |author2=K. D. Jordan | title = Comparison of Density Functional and MP2 Calculations on the Water Monomer and Dimer | journal = J. Phys. Chem. | volume = 98 | issue = 40 | pages = 10089–10094 | year = 1994 | doi = 10.1021/j100091a024 }}
4. ^{{ cite journal |author1=P.J. Stephens |author2=F. J. Devlin |author3=C. F. Chabalowski |author4=M. J. Frisch | title = Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields | journal = J. Phys. Chem. | volume = 98 | pages = 11623–11627 | year = 1994 | doi = 10.1021/j100096a001 | issue = 45 }}
5. ^{{cite journal | author = A. D. Becke | title = Density-functional exchange-energy approximation with correct asymptotic behavior | journal = Phys. Rev. A | volume = 38 | pages = 3098–3100 | year = 1988 | url = http://link.aps.org/abstract/PRA/v38/p3098 | doi = 10.1103/PhysRevA.38.3098 | pmid = 9900728 | issue = 6 |bibcode = 1988PhRvA..38.3098B }}
6. ^{{cite journal|author1=Chengteh Lee |author2=Weitao Yang |author3=Robert G. Parr |title=Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density|journal=Phys. Rev. B|volume=37|pages=785–789|year=1988|doi=10.1103/PhysRevB.37.785|bibcode = 1988PhRvB..37..785L|issue= 2 }}
7. ^{{cite journal|title = Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis|author1=S. H. Vosko |author2=L. Wilk |author3=M. Nusair |journal = Can. J. Phys.|volume = 58|pages = 1200–1211|year = 1980|doi = 10.1139/p80-159|bibcode = 1980CaJPh..58.1200V|issue = 8 }}
8. ^{{cite journal|last=Becke|first=Axel D.|year=1993|title=Density-functional thermochemistry. III. The role of exact exchange|journal=J. Chem. Phys.|volume=98|issue=7|pages=5648–5652|doi=10.1063/1.464913 |bibcode = 1993JChPh..98.5648B }}
9. ^{{cite journal| doi = 10.1063/1.478522| issn = 0021-9606| volume = 110| issue = 13| pages = 6158–6170| last = Adamo| first = Carlo|author2=Vincenzo Barone| title = Toward reliable density functional methods without adjustable parameters: The PBE0 model| journal = The Journal of Chemical Physics| date = 1999-04-01|bibcode = 1999JChPh.110.6158A }}
10. ^1 {{ cite journal | doi = 10.1103/PhysRevLett.77.3865| volume = 77 | issue = 18 | pages = 3865–3868 | last = Perdew | first = John P. |author2=Kieron Burke |author3=Matthias Ernzerhof | title = Generalized Gradient Approximation Made Simple | journal = Physical Review Letters | date = 1996-10-28 | pmid=10062328|bibcode = 1996PhRvL..77.3865P }}
11. ^{{ cite journal |author1=Jochen Heyd |author2=Gustavo E. Scuseria |author3=Matthias Ernzerhof | title = Hybrid functionals based on a screened Coulomb potential | journal = J. Chem. Phys. | volume = 118 | issue = 18 | pages = 8207 | year = 2003 | doi = 10.1063/1.1564060 |bibcode = 2003JChPh.118.8207H }}
12. ^{{ cite journal | title=The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals | doi = 10.1007/s00214-007-0310-x| volume = 120 | issue = 1–3| pages = 215–241 | last = Zhao | first = Yan |author2=Donald G. Truhlar | journal = Theor. Chem. Account | year = 2008}}
13. ^{{ cite journal | title=Density Functional for Spectroscopy: No Long-Range Self-Interaction Error, Good Performance for Rydberg and Charge-Transfer States, and Better Performance on Average than B3LYP for Ground States | doi = 10.1021/jp066479k | pmid = 17149824 | volume = 110 | issue = 49 | pages = 13126–13130 | last = Zhao | first = Yan |author2=Donald G. Truhlar | journal = J. Phys. Chem. | year = 2006 }}
- 1993–94 Meistriliiga
- 1993–94 Mersin Idmanyurdu season
- 1993–94 Mersin İdmanyurdu season
- 1993–94 MetJHL season
- 1993–94 Mexican Primera División season
- 1993–94 Miami Heat season
- 1993–94 Michigan Wolverines men's basketball team
- 1993–94 Mighty Ducks of Anaheim season
- 1993–94 Minnesota North Stars season
- 1993–94 Moldovan National Division
- 1993–94 Montreal Canadiens season
- 1993–94 Montréal Canadiens season
- 1993–94 National Hurling League
- 1993–94 Nationalliga A
- 1993–94 National Professional Soccer League season
- 1993–94 National Soccer League
- 1993–94 Nemzeti Bajnoksag I
- 1993–94 Nemzeti Bajnokság I
- 1993–94 Newcastle United F.C. season
- 1993–94 Newport A.F.C. season
- 1993–94 Newport County A.F.C. season
- 1993–94 New York Islanders season
- 1993–94 New York Knicks season
- 1993–94 NOFV-Oberliga
- 1993–94 North Carolina Tar Heels women's basketball team