直角梯形zhijiao tixing
有一腰垂直于底的梯形.如图,在梯形ABCD中,AB∥DC,CB⊥AB,ABCD是直角梯形.直角梯形垂直于底的腰上的两个角是直角.在梯形中,最多有两个角是直角.直角梯形经常被分割为一个矩形和一个直角三角形加以研究.
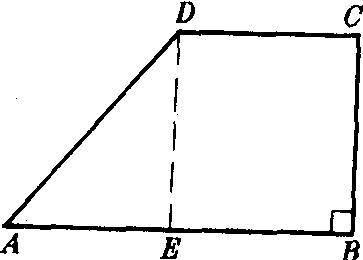
| 词条 | 直角梯形 |
| 类别 | 中文百科知识 |
| 释义 | 直角梯形zhijiao tixing有一腰垂直于底的梯形.如图,在梯形ABCD中,AB∥DC,CB⊥AB,ABCD是直角梯形.直角梯形垂直于底的腰上的两个角是直角.在梯形中,最多有两个角是直角.直角梯形经常被分割为一个矩形和一个直角三角形加以研究.
|
| 随便看 |
开放百科全书收录579518条英语、德语、日语等多语种百科知识,基本涵盖了大多数领域的百科知识,是一部内容自由、开放的电子版国际百科全书。