- Definition
- Background Sequence alignment Distance matrix method
- The problem of tree alignment
- Combinatorial optimization strategy The keyword tree theory and the Aho-Corasick search algorithm Keyword tree theory Aho-Corasick search algorithm
- Other strategies Heuristic algorithm Tree alignment graph
- See also
- References
In computational phylogenetics, tree alignment is a computational problem concerned with producing multiple sequence alignments, or alignments of three or more sequences of DNA, RNA, or protein. Sequences are arranged into a phylogenetic tree, modeling the evolutionary relationships between species or taxa. The edit distances between sequences are calculated for each of the tree's internal vertices, such that the sum of all edit distances within the tree is minimized. Tree alignment can be accomplished using one of several algorithms with various trade-offs between manageable tree size and computational effort.
Definition
Input: A set  of sequences, a phylogenetic tree
of sequences, a phylogenetic tree 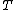 leaf-labeled by and an edit distance function
leaf-labeled by and an edit distance function  between sequences.
between sequences.
Output: A labeling of the internal vertices of such that 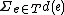 is minimized, where
is minimized, where 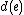 is the edit distance between the endpoints of
is the edit distance between the endpoints of  .
.
The task is NP-hard.[1]
Background
Sequence alignment
{{Main|Sequence alignment}}In bioinformatics, the basic method of information processing is to contrast the sequence data. Biologists use it to discover the function, structure, and evolutionary information in biological sequences. The following analyses are based on the sequence assembly: the phylogenetic analysis, the haplotype comparison, and the prediction of RNA structure. Therefore, the efficiency of sequence alignment will directly affect the efficacy of solving these problems. In order to design a rational and efficient sequence alignment, the algorithm derivation becomes an important branch of research in the field of bioinformatics.
Generally, sequence alignment means constructing a string from two or more given strings with the greatest similarity by adding letters, deleting letters, or adding a space for each string. The multiple sequence alignment problem is generally based on pairwise sequence alignment and currently, for a pairwise sequence alignment problem, biologists can use a dynamic programming approach to obtain its optimal solution. However, the multiple sequence alignment problem is still one of the more challenging problems in bioinformatics. This is because finding the optimal solution for multiple sequence alignment has been proven as an NP-complete problem and only an approximate optimal solution can be obtained.[2]
Distance matrix method
{{Main|Distance matrix method distance}}Distance method measures the minimum operation number of character insertions, deletions, and substitutions that are required to transform one sequence u to the other sequence v when being operated on a pair of strings. The calculation of edit distance can be based on dynamic programming, and the equation is in O(|u|×|v|) time, where |u| and |v| are the lengths of u and v.[3] The efficient estimation of edit distance is essential as Distance method is a basic principle in computational biology [4] For functions of hereditary properties “symmetrization” can be used. Due to a series of functions being used to calculate edit distance, different functions may result in different results. Finding an optimal edit distance function is essential for the tree alignment problem.
The problem of tree alignment
{{Unreferenced section|date=January 2019}}Tree alignment results in a NP-hard problem, where scoring modes and alphabet sizes are restricted. It can be found as an algorithm, which is used to find the optimized solution. However, there is an exponential relationship between its efficiency and the number sequences, which means that when the length of the sequence is very large, the computation time required to get results is enormously long. Using star alignment to get the approximate optimized solution is faster than using tree alignment. However, whatever the degree of multiple-sequence similarity is, the time complexity of star alignment has a proportional relationship with the square of the sequence number and the square of the sequence average length. As usual, the sequence in MSA is so long that it is also inefficient or even unacceptable. Therefore, the challenge of reducing the time complexity to linear is one of the core issues in tree alignment.
Combinatorial optimization strategy
Combinatorial optimization is a good strategy to solve MSA problems. The idea of combinatorial optimization strategy is to transform the multiple sequence alignment into pair sequence alignment to solve this problem. Depending on its transformation strategy, the combinatorial optimization strategy can be divided into the tree alignment algorithm and the star alignment algorithm. For a given multi-sequence set ={ ,…,
,…,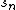 }, find an evolutionary tree which has n leaf nodes and establishing one to one relationship between this evolutionary tree and the set . By assigning the sequence to the internal nodes of the evolutionary tree, we calculate the total score of each edge, and the sum of all edges' scores is the score of the evolutionary tree. The aim of tree alignment is to find an assigned sequence, which can obtain a maximum score, and get the final matching result from the evolutionary tree and its nodes' assigned sequence. Star alignment can be seen as a special case of the tree alignment. When we use star alignment, the evolutionary tree has only one internal node and n leaf nodes. The sequence, which is assigned to the internal node, is called the core sequence.[5]
}, find an evolutionary tree which has n leaf nodes and establishing one to one relationship between this evolutionary tree and the set . By assigning the sequence to the internal nodes of the evolutionary tree, we calculate the total score of each edge, and the sum of all edges' scores is the score of the evolutionary tree. The aim of tree alignment is to find an assigned sequence, which can obtain a maximum score, and get the final matching result from the evolutionary tree and its nodes' assigned sequence. Star alignment can be seen as a special case of the tree alignment. When we use star alignment, the evolutionary tree has only one internal node and n leaf nodes. The sequence, which is assigned to the internal node, is called the core sequence.[5]The keyword tree theory and the Aho-Corasick search algorithm
When the combinatorial optimization strategy is used to transform the multiple sequence alignment into pair sequence alignment, the main problem is changed from "How to improve the efficiency of multiple sequence alignment" to "How to improve the efficiency of pairwise sequence alignment." The Keyword Tree Theory and the Aho-Corasick search algorithm is an efficient approach to solve the pairwise sequence alignment problem. The aim of combining the keyword tree theory and the Aho-Corasick search algorithm is to solve this kind of problem: for a given long string and a set of short strings  ={
={ ,
, ,… ,
,… ,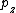 } (z∈N,z>1), find the location of all
} (z∈N,z>1), find the location of all  in . The keyword tree produced by set is used, and then searched for in with this keyword tree through the Aho-Corasick search algorithm.[6] The total time complexity of using this method to find all ’s location in the T is O(
in . The keyword tree produced by set is used, and then searched for in with this keyword tree through the Aho-Corasick search algorithm.[6] The total time complexity of using this method to find all ’s location in the T is O( +
+ +
+ ), where =|| (the length of ), =∑|| (the sum of all 's lengths) and means the sum of occurrence for all in .
), where =|| (the length of ), =∑|| (the sum of all 's lengths) and means the sum of occurrence for all in .
Keyword tree theory
The keyword tree of the setm ={,,… , } (z∈N,z>1) mis a rooted tree, whose root denoted by K, and this keyword tree satisfies:
(1): Each edge clearly demarcates one letter.
(2): Any two edges separated from the same node are to correspond to different letters.
(3) Each pattern (i=1,2,…,z) corresponds to a node  , and the path from the root K to the node can exactly correctly spell the string .
, and the path from the root K to the node can exactly correctly spell the string .
For each leaf node of this K tree, it corresponds to one of the certain patterns of set .
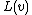 is used to represent the STRING which is connected from the root node to the node .
is used to represent the STRING which is connected from the root node to the node .  will then be used to represent the length of the longest suffix (also, this suffix is the prefix of one of patterns in the set ). Searching this prefix from the root node in the keyword tree, and the last node denoted by
will then be used to represent the length of the longest suffix (also, this suffix is the prefix of one of patterns in the set ). Searching this prefix from the root node in the keyword tree, and the last node denoted by 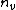 when the search is over.{{clarify|date=January 2019|reason=unclear phrasing, "Searching this prefix from the root node in the keyword tree, and the last node denoted by n_v when the search is over. When Lp(v)=0, n_v =K. The ordered pair (v, n_v) is called a failure link." This is unclear, and I don't know what it means.}}[7]
when the search is over.{{clarify|date=January 2019|reason=unclear phrasing, "Searching this prefix from the root node in the keyword tree, and the last node denoted by n_v when the search is over. When Lp(v)=0, n_v =K. The ordered pair (v, n_v) is called a failure link." This is unclear, and I don't know what it means.}}[7]For example, the set ={potato, tattoo, theater, other}, and the keyword tree is shown on the right. In that example, if =potat, then =|tat|=3, and the failure link of the node is shown in that figure.
Establishing a failure link is the key to improve the time complexity of the Aho-Corasick algorithm. It can be used to reduce the original polynomial time to the linear time for searching. Therefore, the core of keyword tree theory is to find all failure links (which also means finding all s) of a keyword tree in the linear time. It is assumed that every of all nodes , whose distance from the root node is less than or equal , can be found. The of the node whose distance from the root node is + 1 can then be sought for. Its parent node is 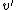 , and the letter represented by the node and , is
, and the letter represented by the node and , is  .
.
(1): If the next letter of the node 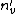 is , the other node of this edge can be set as
is , the other node of this edge can be set as  , and =.
, and =.
(2): If all letters are not by searching all edges between and its child nodes, 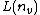 is a suffix of
is a suffix of 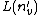 plus . Because this suffix matches the STRING beginning with the root node (similar to prefix), the after can be detected or not. If not, this process can be continued until or the root node is found.
plus . Because this suffix matches the STRING beginning with the root node (similar to prefix), the after can be detected or not. If not, this process can be continued until or the root node is found.
Aho-Corasick search algorithm
{{Main|Aho–Corasick string matching algorithm}}After establishing all failure links in the keyword tree, the Aho-Corasick search algorithm is used to find the locations of all (i=1,2,…,z) in the linear time. In this step, the time complexity is O(m+k).
Other strategies
{{Main|Multiple sequence alignment}}In MSA, DNA, RNA, and proteins, sequences are usually generated and they are assumed to have an evolutionary relationship. By comparing generated maps of RNA, DNA, and sequences from evolutionary families, people can assess conservation of proteins and find functional gene domains by comparing differences between evolutionary sequences. Generally, heuristic algorithms and tree alignment graphs are also adopted to solve multiple sequence alignment problems.
Heuristic algorithm
{{Main|Heuristic (computer science)}}Generally, heuristic algorithms rely on the iterative strategy, which is to say that based on a comparison method, optimizing the results of multiple sequence alignment by the iterative process. Davie M. proposed using the particle swarm optimization algorithm to solve the multiple sequence alignment problem; Ikeda Takahiro proposed a heuristic algorithm which is based on the A* search algorithm; E. Birney first proposed using the hidden Markov model to solve the multiple sequence alignment problem; and many other biologists use the genetic algorithm to solve it.[8][9] All these algorithms generally are robust and insensitive to the number of sequences, but they also have shortcomings. For example, the results from the particle swarm optimization algorithm are unstable and its merits depend on the selection of random numbers, the runtime of the A* search algorithm is too long, and the genetic algorithm is easy to fall into local excellent.{{clarify|date=January 2019|reason=what does "local excellent" mean?}}
Tree alignment graph
Roughly, tree alignment graph aims to align trees into a graph and finally synthesize them to develop statistics. In biology, tree alignment graphs (TAGs) are used to remove the evolutionary conflicts or overlapping taxa from sets of trees and can then be queried to explore uncertainty and conflict. By integrating methods of aligning, synthesizing and analyzing, the TAG aims to solve the conflicting relationships and partial overlapping taxon sets obtained from a wide range of sequences. Also, the tree alignment graph serves as a fundamental approach for supertree and grafting exercises, which have been successfully tested to construct supertrees by Berry.[10] Because the transformation from trees to a graph contain similar nodes and edges from their source trees, TAGs can also provide extraction of original source trees for further analysis. TAG is a combination of a set of aligning trees. It can store conflicting hypotheses evolutionary relationship and synthesize the source trees to develop evolutionary hypotheses. Therefore, it is a basic method to solve other alignment problems.[11]
See also
- Generalized tree alignment
References
2. ^L Wang, T Jiang. On the complexity of multiple sequence alignment[J]. Journal of Computational Biology, 194,1(4):337— 34.
3. ^Yen Hung Chen, On the bottleneck tree alignment problems, INFORMATION SCIENCES; JUN 1, 2010; 180; 11; p2134-p2141
4. ^{{cite journal | last=Ostrovsky | first=Rafail | last2=Rabani | first2=Yuval | title=Low distortion embeddings for edit distance | journal=Journal of the ACM | publisher=Association for Computing Machinery (ACM) | volume=54 | issue=5 | date=2007-10-01 | issn=0004-5411 | doi=10.1145/1284320.1284322 | pages=23–es | ref=harv}}
5. ^Serafim Batzoglou. The many faces of sequence alignment[J]. Briefings in Bioinformatics. 2005,6(1):6—22
6. ^Aho A V, Corasick M J. Efficient string matching: an aid to bibliographic search[J]. Communications of ACM, 1975,18(6): 333—340{{Dead link|date=July 2018 |bot=InternetArchiveBot |fix-attempted=no }}.
7. ^D Gusfield. Algorithms on strings, trees and sequences: computer science and computational biology[M]. Cambridge: Cambridge University Press.1997.
8. ^RobertC Edgar, Serafim Batzoglou. Multiple sequence alignment[J]. Current Opinion in Structural Biology. 2006,16(3):368— 373 {{Webarchive|url=https://web.archive.org/web/20131023062245/http://robotics.stanford.edu/~serafim/Publications/2006_MSA_COSB.pdf |date=2013-10-23 }}.
9. ^Notredame C, Higgins D.G. SAGA:sequence alignment by genetic algorithm [J]. Nucleic Acids Research. 1996,24(8):1515-1524.
10. ^Wilkinson M, Pisani D, Measuring support and finding unsupported relationships in supertrees, Systematic Biology 54:823-831.
11. ^Stephen A. Smith, Joseph W. Brown, analyzing and synthesizing phylogenies using tree alignment graphs, PLoS Computational Biology 9(9).
- Cabriite
- Cabril Amilcar
- Cabrillanes
- Cabrillanes, León
- Cabrillanes, Spain
- Cabrillas
- Cabrillo Beach
- Cabrillo Bridge
- Cabrillo Elementary School
- Cabrillo, Estévan
- Cabrillo High School (Lompoc, California)
- Cabrillo High School (Long Beach)
- Cabrillo High School (Long Beach, CA)
- Cabrillo Highway
- Cabrillo, Juan Rodríguez
- Cabrillo Marine Aquarium
- Cabrils
- Cabrini Boulevard
- Cabrini High School
- Cabrini High School (disambiguation)
- Cabrini High School for Girls
- Cabrini High School (Michigan)
- Cabrini High School (New Orleans)
- Cabrio
- Cabrio coaches