- History
- Theory
- Applications Cycloadditions Sigmatropic reactions Electrocyclic reactions
- See also
- References
In chemistry, frontier molecular orbital theory is an application of MO theory describing HOMO/LUMO interactions.
History
In 1952, Kenichi Fukui published a paper in the Journal of Chemical Physics titled "A molecular theory of reactivity in aromatic hydrocarbons."[1] Though widely criticized at the time, he later shared the Nobel Prize in Chemistry with Roald Hoffmann for his work on reaction mechanisms. Hoffman's work focused on creating a set of four pericyclic reactions in organic chemistry, based on orbital symmetry, which he coauthored with Robert Burns Woodward, entitled "The Conservation of Orbital Symmetry."
Fukui's own work looked at the frontier orbitals, and in particular the effects of the Highest Occupied Molecular Orbital (HOMO) and the Lowest Unoccupied Molecular Orbital (LUMO) on reaction mechanisms, which led to it being called Frontier Molecular Orbital Theory (FMO Theory). He used these interactions to better understand the conclusions of the Woodward–Hoffmann rules.
Theory
Fukui realized that a good approximation for reactivity could be found by looking at the frontier orbitals (HOMO/LUMO). This was based on three main observations of molecular orbital theory as two molecules interact:
- The occupied orbitals of different molecules repel each other.
- Positive charges of one molecule attract the negative charges of the other.
- The occupied orbitals of one molecule and the unoccupied orbitals of the other (especially the HOMO and LUMO) interact with each other causing attraction.
In general, the total energy change of the reactants on approach of the transition state is described by the Klopman-Salem equation, derived from perturbational MO theory. The first and second observations correspond to taking into consideration the filled-filled interaction and Coulombic interaction terms of the equation, respectively. With respect to the third observation, primary consideration of the HOMO-LUMO interaction is justified by the fact that the largest contribution in the filled-unfilled interaction term of the Klopman-Salem equation comes from molecular orbitals r and s that are closest in energy (i.e., smallest 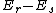 value).[2] From these observations, frontier molecular orbital (FMO) theory simplifies prediction of reactivity to analysis of the interaction between the more energetically matched HOMO-LUMO pairing of the two reactants. In addition to providing a unified explanation of diverse aspects of chemical reactivity and selectivity, it agrees with the predictions of the Woodward–Hoffmann orbital symmetry and Dewar-Zimmerman aromatic transition state treatments of thermal pericyclic reactions, which are summarized in the following selection rule:
value).[2] From these observations, frontier molecular orbital (FMO) theory simplifies prediction of reactivity to analysis of the interaction between the more energetically matched HOMO-LUMO pairing of the two reactants. In addition to providing a unified explanation of diverse aspects of chemical reactivity and selectivity, it agrees with the predictions of the Woodward–Hoffmann orbital symmetry and Dewar-Zimmerman aromatic transition state treatments of thermal pericyclic reactions, which are summarized in the following selection rule:
"A ground-state pericyclic change is symmetry-allowed when the total number of (4q+2)s and (4r)a components is odd"
(4q+2)s refers to the number of aromatic, suprafacial electron systems; likewise, (4r)a refers to antiaromatic, antarafacial systems. It can be shown that if the total number of these systems is odd then the reaction is thermally allowed.[2]
Applications
Cycloadditions
A cycloaddition is a reaction that simultaneously forms at least two new bonds, and in doing so, converts two or more open-chain molecules into rings.[3] The transition states for these reactions typically involves the electrons of the molecules moving in continuous rings, making it a pericyclic reaction. These reactions can be predicted by the Woodward–Hoffmann rules and thus are closely approximated by FMO Theory.
The Diels–Alder reaction between maleic anhydride and cyclopentadiene is allowed by the Woodward–Hoffmann rules because there are six electrons moving suprafacially and no electrons moving antarafacially. Thus, there is one (4q + 2)s component and no (4r)a component, which means the reaction is allowed thermally.
FMO theory also finds that this reaction is allowed and goes even further by predicting its stereoselectivity, which is unknown under the Woodward-Hoffmann rules. Since this is a [4 + 2], the reaction can be simplified by considering the reaction between butadiene and ethene. The HOMO of butadiene and the LUMO of ethene are both antisymmetric (rotationally symmetric), meaning the reaction is allowed.*
In terms of the stereoselectivity of the reaction between maleic anhydride and cyclopentadiene, the endo-product is favored, a result best explained through FMO theory. The maleic anhydride is an electron-withdrawing species that makes the dieneophile electron deficient, forcing the regular Diels–Alder reaction. Thus, only the reaction between the HOMO of cyclopentadiene and the LUMO of maleic anhydride is allowed. Furthermore, though the exo-product is the more thermodynamically stable isomer, there are secondary (non-bonding) orbital interactions in the endo- transition state, lowering its energy and making the reaction towards the endo- product faster, and therefore more kinetically favorable. Since the exo-product has primary (bonding) orbital interactions it can still form, but since the endo-product forms faster it is the major product.[2]
*Note: The HOMO of ethene and the LUMO of butadiene are both symmetric, meaning the reaction between these species is allowed as well. This is referred to as the "inverse electron demand Diels–Alder."Sigmatropic reactions
A sigmatropic rearrangement is a reaction in which a sigma bond moves across a conjugated pi system with a concomitant shift in the pi bonds. The shift in the sigma bond may be antarafacial or suprafacial. In the example of a [1,5] shift in pentadiene, if there is a suprafacial shift, there is 6 e− moving suprafacially and none moving antarafacially, implying this reaction is allowed by the Woodward–Hoffmann rules. For an antarafacial shift, the reaction is not allowed.
These results can be predicted with FMO theory by observing the interaction between the HOMO and LUMO of the species. To use FMO theory, the reaction should be considered as two separate ideas: (1) whether or not the reaction is allowed, and (2) which mechanism the reaction proceeds though. In the case of a [1,5] shift on pentadiene, the HOMO of the sigma bond (i.e. a constructive bond) and the LUMO of butadiene on the remaining 4 carbons is observed. Assuming the reaction happens suprafacially, the shift results with the HOMO of butadiene on the 4 carbons that are not involved in the sigma bond of the product. Since the pi system changed from the LUMO to the HOMO, this reaction is allowed (though it would not be allowed if the pi system went from LUMO to LUMO).
To explain why the reaction happens suprafacially, first notice that the terminal orbitals are in the same phase. For there to be a constructive sigma bond formed after the shift, the reaction would have to be suprafacial. If the species shifted antarafacially then it would form an antibonding orbital and there would not be a constructive sigma shift.
It is worth noting that in propene the shift would have to be antarafacial, but since the molecule is very small that twist is not possible and the reaction is not allowed.
Electrocyclic reactions
An electrocyclic reaction is a pericyclic reaction involving the net loss of a pi bond and creation of a sigma bond with formation of a ring. This reaction proceeds through either a conrotatory or disrotatory mechanism. In the conrotatory ring opening of cyclobutene, there are two electrons moving suprafacially (on the pi bond) and two moving antarafacially (on the sigma bond). This means there is one 4q + 2 suprafacial system and no 4r antarafacial system; thus the conrotatory process is thermally allowed by the Woodward–Hoffmann rules.
The HOMO of the sigma bond (i.e. a constructive bond) and the LUMO of the pi bond are important in the FMO theory consideration. If the ring opening uses a conrotatory process then the reaction results with the HOMO of butadiene. As in the previous examples the pi system moves from a LUMO species to a HOMO species, meaning this reaction is allowed.[2]
See also
- Addition to pi ligands
- Klopman-Salem equation
- Oxy cope elimination pericyclic reaction
References
2. ^1 2 3 {{cite book|title=Frontier Orbitals and Organic Chemical Reactions|last=Fleming|first=Ian|publisher=Wiley|year=1978|isbn=0-471-01819-8|location=London|pages=24–109|authorlink=Ian Fleming (chemist)}}
3. ^{{cite book | last = Miller | first = Bernard | title = Advanced Organic Chemistry: Reactions and Mechanisms | publisher = Pearsons | year = 2004 | location = Upper Saddle River, NJ | pages = 53–54 | isbn = 0-13-065588-0}}
- Bedrock, CO
- Bedrock gardens
- Bedrock Gardens
- Bedrock plane
- Bedrock river
- Bedrock shiner
- Bedrock (song)
- Bedrock song
- Bedrock Studios
- Bedrock Vice
- Bedrock (Young Money song)
- Bedroom bondage
- Bedroom Boogie
- Bedroom Boom
- Bedroom Bound
- Bedroom Bound EP
- Bedroom Classics, Vol. 3
- Bedroom Eyes
- Bedroom Eyes (disambiguation)
- Bedroom Eyes (film)
- Bedroom Eyes (movie)
- Bedroom Eyes (musician)
- Bedroom Eyes (song)
- Bedroom in arles
- Bedroom philosopher